We have added a page on the pituitary gland
Fabry as a lysosomal storage disease, is there possibly more to it?
Yes, at least in a certain proportion of patients.
There is always talk of an enzyme deficiency that leads to deposits in the cells and then triggers the malfunctions. In many missense variants (i.e. an amino acid is altered somewhere in the DNA on the GLA gene) there is a residual activity of the enzyme. Nevertheless, the patients are often also severely affected. Here, another mechanism comes into play: ER stress or Unfolded Protein Response (UPR).
A brief explanation: Due to the variant, the enzyme is formed, but it is partly defective. Now a kind of “quality assurance” kicks in in the cell. The faulty enzyme is “repaired”. This is a normal process in the cells, since minor errors can happen again and again. However, due to the variant, the enzyme is permanently formed incorrectly and the error correction is overloaded. This is ER stress, which can lead to malfunctions of a cell or even cause cell death.
Caperone therapy also “repairs” the wrong enzyme and relieves the cell. Here, enzyme replacement therapy would be less effective.
This mechanism has been demonstrated for some time, including for some of the controversial variants. But this explanation is not accepted by all scientists because it contradicts their own publications.
We patients are the ones who suffer; we are then prescribed no therapy or a less effective one.
Genetic testing, what and how is actually tested…
In Fabry and many other diseases, a genetic test is used to determine whether there are changes in the DNA (genetic material) that can cause a disease.
For some time now, “next generation sequencing” has been available for this purpose, i.e. the actual sequence of information on a gene is determined. In the past, this was not possible and special tests were developed that were applied to very specific gene sections (e.g. MLPA analysis). However, this can only detect a part of the changes (only deletions and duplications). Since most of the Fabry mutations known so far are missense mutations (exchange of a base pair in the DNA), this simple test cannot detect Fabry in most cases.
A complete analysis can only be carried out using “Next Generation Sequencing” (NGS for short). It must be ensured that intronic regions are also analysed, details of which we have already described here.
An overview of different test methods can be found here.
Unfortunately, there are information deficits here as well. We have a negative Fabry gene test which is only based on a simple MLPA analysis and therefore has almost no significance. In the information on the test used, the limitations are clearly explained, yet for the examining geneticist, Fabry was ruled out at that point. This is another piece of the puzzle as to why the diagnosis of rare diseases takes so long. Wrong tools are used by even professionals and so wrong diagnoses are made.
Fabry misdiagnoses: MS, ME/CFS, fibromyalgia, Parkinson’s disease, and more.

Frequent misdiagnoses result in a Fabry diagnosis taking over 15 years on average. Many patients are never properly diagnosed. However, many of them could be helped causally. We have listed all known misdiagnoses here.
What are intronic mutations and why do they make people ill?
We are often contacted by patients with Fabry-related conditions who have intronic mutations in their genetics. Therefore, we would like to turn our and your attention back to intronic mutations:
Intronic mutations are an even broader topic. If you ask the Fabry experts, they usually explain that intronic mutations cannot do anything because there is no information in the introns. The answer is that simple and that wrong.
If you want to read the details and have access to Pubmed: https://pubmed.ncbi.nlm.nih.gov/28497172/ (“Deep intronic mutations and human disease”, direct at Springer: https://link.springer.com/article/10.1007/s00439-017-1809-4, Human Genetics volume 136, pages 1093–1111; 2017)
Briefly and simplified: on the one hand, there is also regulatory information in the introns that has an influence on enzyme production. On the other hand, certain sequences of the DNA sequence are used to determine between the introns and exons. If a new exon start is created by mutation in the intron, then the gene is read incorrectly (even if the “original” exons are all correct).
Unfortunately, most gene tests do not pay attention to intronic mutations at all, these areas are not analysed at all. But at the latest, if lyso-GB3-values or enzyme activity are abnormal, one has to search for the cause, which can then lie in the intron.
See also the article on intronic mutations on our website.
Study situation: where does the “proof” of the apathogenicity of D313Y and other controversial mutations come from?
Various publications attempt to prove the apathogenicity of the controversial mutations. Some of them describe rather individual cases and are therefore not statistically reliable. Others refer to previous publications when making their statements. If you now look at the sources where the original statements with evidence come from, you will very often find two studies from 2003:
- Yasuda: “Fabry disease: characterisation of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele”.
- Froissart: “Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma”.
Both authors worked closely together, i.e. the studies are not independent! The studies show altered biochemical and physical properties of the modified enzyme. However, they then assume that these changes have no effect on the cellular level. But already in this study, disturbed processes in the cells are detected, which were later further investigated in studies on another controversial mutation. There it was shown that these changes lead to so-called ER stress (ER = endoplasmic reticulum). This ER stress is the cause of cell malfunction and even cell death. (see also Unfolded Protein Response)
Another point is the so-called ER stress, more about this in our blog entry:
Fabry as a lysosomal storage disease, is there possibly more to it.
(Source e.g. https://doi.org/10.1101/2022.09.27.509714)
I.e. the statements on apathogenicity are often not reliable or even wrong.
Therefore, most authors add that due to the small number of cases and various open questions, apathogenicity cannot necessarily be proven and further research is necessary.
Important Research that is made impossible by the deliberate filtering out of the controversial mutations in the gene laboratory.
Example:
A publication from 2020 also refers to these two studies to prove apathogenicity.
In this publication (“An expert consensus document on the management of cardiovascular manifestations of Fabry disease”, Linhart et al. 2020) it was written: “As a cautionary example, the p.Asp313Tyr change results in a serum pseudodeficiency of AGAL-A activity and is not disease-causing. Similarly, a number of GLA variants previously thought to be disease-causing (e.g. p.Arg118Cys) have been shown to be of uncertain significance or likely benign. “(This is followed by a citation)
We have traced back the sources until we reached the original source:
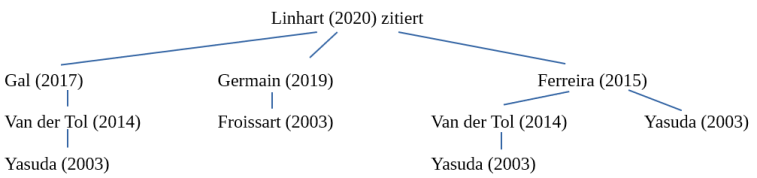
In turn, the Varsome database cites Yasuda’s study as a source to prove pathogenicity.
Who determines whether a genetic variant is pathogenic or not?
There are five classifications regarding the pathogenicity of mutations/variants:
- pathogenic – causing disease
- likely pathogenic – probably causing disease
- uncertain significance
- likely benign
- benign
In Germany the Genetic Diagnostics Commission has decided that laboratories should only report pathogenic and likely pathogenic variants in their findings. Variants of unclear significance can be mentioned in the findings. Benign and probably benign variants should no longer be reported.
Since there are a large number of benign variants in genetics, it seems perfectly understandable to filter them out.
But who determines whether a variant is pathogenic or not? And what about the variants where scientists do not agree (=unclear significance)? And who does the classification?
The classification can and must be done by each laboratory/doctor/centre itself. There are ACMG (American College of Medical Genetics) guidelines for this. However, these guidelines are under discussion, e.g. for Fabry itself. They also leave considerable room for interpretation. For example, the D313Y variant is classified by laboratories as “likely benign”. With other studies as a reference, this variant can also be classified as “likely pathogenic” (see e.g. Varsome database).
The literature situation is currently inconclusive; more recent work points to pathogenic factors. Nevertheless, some variants (such as the D313Y or A143T) have been reclassified as benign by some laboratories/centres and are now not even reported.
So, on the one hand, the stipulation of the Genetic Engineering Commission and the decision of doctors and laboratories is incomprehensible, because this actively keeps patients away from diagnosis and therapy. On the other hand, it also impairs research or even prevents it altogether.
We continue to fight for the controversial mutations, but the fight is severely and systematically hindered.
Literature links for interested people
We created a page with literature links und explanations for interested people: Literature links for interested people
Patient reports on her experiences at the geneticist
You can find the article under Laboratory test / Genetics
It is shocking how little is known about Fabry disease, although there has been a therapy for more than 20 years.
Much false information is still widespread:
Starting with the number of people affected. The frequency is given as 1 in 40,000. There are mutations that are found much more frequently in the population. Example the variant p.D313Y. This is estimated to affect 1 in 200 people.
Men are affected more often.
Women are only carriers and would not get sick themselves.
The healthy X in women can compensate for the sick.
Unfortunately, the so-called biomarkers such as measuring α-galactosidase A activity and determining the lyso-Gb3 level cannot provide unambiguous results either, since a gene defect can be present regardless of gender, despite normal values.
Details on variant A143T:
The first patient described by William Anderson in 1898 could be traced back more than a century by family tree. He had the Varainte A143T.
One of his descendants was one of the first men to be treated with enzyme replacement therapy.
Also, the youngest patient starting therapy in the UK at age 3 had variant A143T.
Attribution:
Three Significant Milestones and a Review of the A143T Mutation Within one Family with Anderson Fabry Disease
P. Rohman, Uma Ramaswami, A. Mehta, D.A. Hughes
Royal Free Hospital, Lysosomal Storage Disorders Unit, Pond
St, London, NW3 2QG
52.ERA-EDTA Kongress in London (The European Renal Association and European Dialysis and Transplant Association congress)